
There is little economic incentive to use diagnostic tests before antibiotics. For instance, the empirical treatment of an Uncomplicated Urinary Tract Infection (UTI) with a broad-spectrum antibiotic for 3 to 5 days costs between €4 and €28. In comparison, a uranalysis costs €27 to €73, and culture and susceptibility testing add another 80€ to 180€. Therefore, the commercial potential of an AMR test heavily depends on its cost.
Sadly enough, the In Vitro Diagnostic Regulation (IVDR) has significantly increased the development cost, time-to-market and the risks to get there, with no added value for the developer, the legislator or the patient. The development of many products has been slowed down and even abandoned.
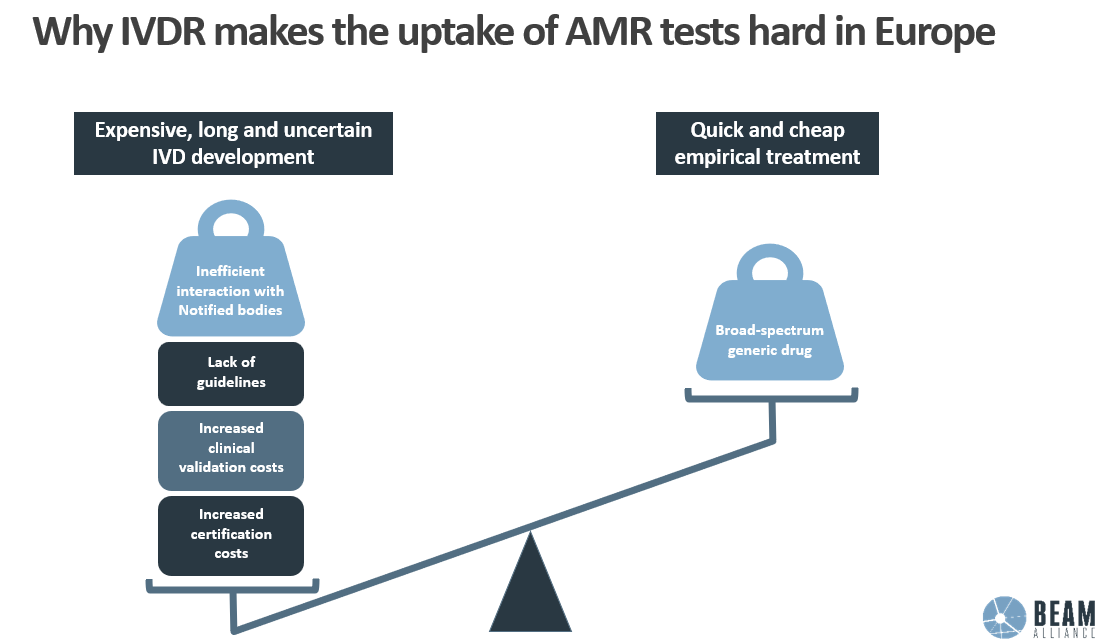
Financial and administrative burden is unbearable for SMEs
- The certification fees exceed €50,000 vs. €6,000 (510k process) or € ~33,000 (De Novo) in the US. The hours spent interpreting the IVDR and the costs hiring additional consultants come on top.
- The clinical validation using performance evaluation studies adds another €150.000 to €300.000.
- The administrative burden increases the work time in the regulatory affairs department by an average of 6 months.
Hidden costs must also be considered, such as those related to waiting the feedback from Notified Bodies.
Altogether, the return on investment can be extended by 6-8 years.
Developers and notified bodies are left on their own
In the US, a test developer can always look up what competitors have done in their own 510k application. This can be very useful for guiding development and saving time. This opportunity is totally missing in the EU, and developers always have to reinvent the wheel: minimal guidelines are needed. In addition, the work of many Notified bodies is criticised: delays, costs, low value for money, low level of understanding of technology (e.g. AI), erroneous interpretation of the regulation, etc. Developers sometimes have the feeling notified bodies establish their own rules, leading to concerning issues of conformity.
How to make things better?
A centralised expertise group is needed, such as FDA’s microbiology department, to define the level of standardisation necessary and sufficient to guarantee an acceptable level of risk, with a risk assessment component.
These recommendations should be enforceable against Notified Bodies. The expert group should therefore speak on behalf of an authoritative organisation, such as a regulatory agency.
A conditional/temporary approval approach where a test can be commercialised based on a minimal set of data and then the large validation study is run with real-world data could help getting users’ feedback.
A review of certification costs should be considered, aligned with costs in the United States.
It is time to be pragmatic and save the EU’s IVD ecosystem.